ElliPro - Tutorial
I. Overview
ElliPro predicts linear and discontinuous antibody epitopes based on a protein antigen's 3D structure.
ElliPro accepts as an input protein structure in PDB format.
If input is a protein sequence, please go to Methods for modeling and docking of antibody and protein 3D structures where
information on methods for modeling and docking of antibody and protein 3D structures is available.
ElliPro associates each predicted epitope with a score, defined as a PI (Protrusion Index) value averaged over epitope residues. In the method, the protein's 3D shape is approximated by a number of ellipsoids, thus that the ellipsoid with PI = 0.9 would include within 90% of the protein residues with 10% of the protein residues being outside of the ellipsoid; while the ellipsoid with PI = 0.8 would include 80% of residues with 20% being outside the ellipsoid. For each residue, a PI value is defined based on the residue's center of mass lying outside the largest possible ellipsoid; for example, all residues that are outside the 90% ellipsoid will have score of 0.9. Residues with larger scores are associated with greater solvent accessibility. Discontinuous epitopes are defined based on PI values and are clustered based on the distance R (in Å between residue's centers of mass). The larger R is associated with the larger discontinues epitopes being predicted. See this paper discussing the method performance and the comparison with other methods.
ElliPro associates each predicted epitope with a score, defined as a PI (Protrusion Index) value averaged over epitope residues. In the method, the protein's 3D shape is approximated by a number of ellipsoids, thus that the ellipsoid with PI = 0.9 would include within 90% of the protein residues with 10% of the protein residues being outside of the ellipsoid; while the ellipsoid with PI = 0.8 would include 80% of residues with 20% being outside the ellipsoid. For each residue, a PI value is defined based on the residue's center of mass lying outside the largest possible ellipsoid; for example, all residues that are outside the 90% ellipsoid will have score of 0.9. Residues with larger scores are associated with greater solvent accessibility. Discontinuous epitopes are defined based on PI values and are clustered based on the distance R (in Å between residue's centers of mass). The larger R is associated with the larger discontinues epitopes being predicted. See this paper discussing the method performance and the comparison with other methods.
II. How to use the tool
Input Page:
- Step 1. Choose an input type: Select "Protein structure ", then go to Step 2b
- Step 2b.
- Specify a protein structure by entering a protein PDB ID or uploading a protein pdb structure from file
- Step 3. Specify epitope prediction parameters (see Epitope prediction parameters)
- Finally Press "Submit"" (Figure 1)
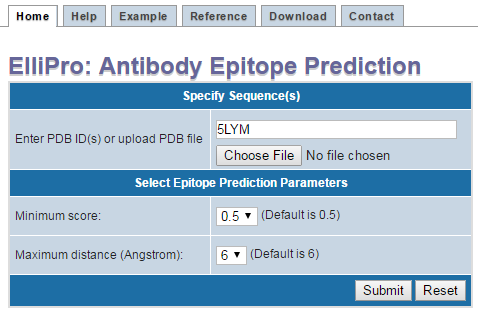
Figure 1 . Example structure input using PDB ID If your protein structure contains more than one chain, ElliPro will ask you to select which chain(s) to be used for epitope prediction. Select one or more chain(s), then click "Submit" (Figure 2 ). IMPORTANT NOTE: ElliPro predicts epitopes for each PDB chain independently, even if more than one chain was selected. Therefore, be cautious when interpreting the results, as the predicted epitope might be buried in the interface with other chains and be solvent unaccessible. If you want to predict epitopes in a multi-chain protein, for example, considering the whole oligomer of the hemoglobin (PDB ID 4HHB, chains A, B, C, D all together), you need to modify a correspondent PDB file (4HHB.pdb) so that all chains of interest have the same chain ID (specified in the column 22 of the ATOM record in the PDB format file). To avoid conflict of residues having the same numbers in the modified PDB file, residues might need to be renumbered.
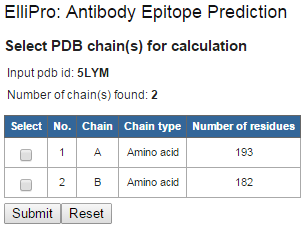
Figure 2 . Example structure input using PDB ID
- Specify "Minimum score" for epitope prediction
- Higher score will predict fewer epitopes and vice versa
- Specify "Maximum distance" for predicting (grouping) discontinous epitopes
- Longer distance will predict discontinuous epitopes that span larger regions and vice vers
III. How prediction results are presented
An example layout of prediction results is shown in Figure 3.
The results show: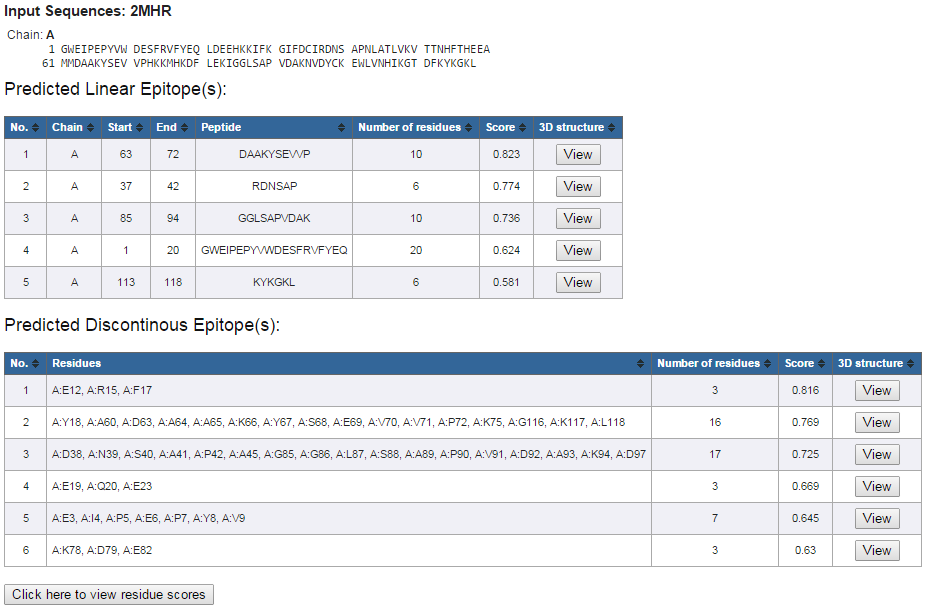
Figure 3. Example prediction results
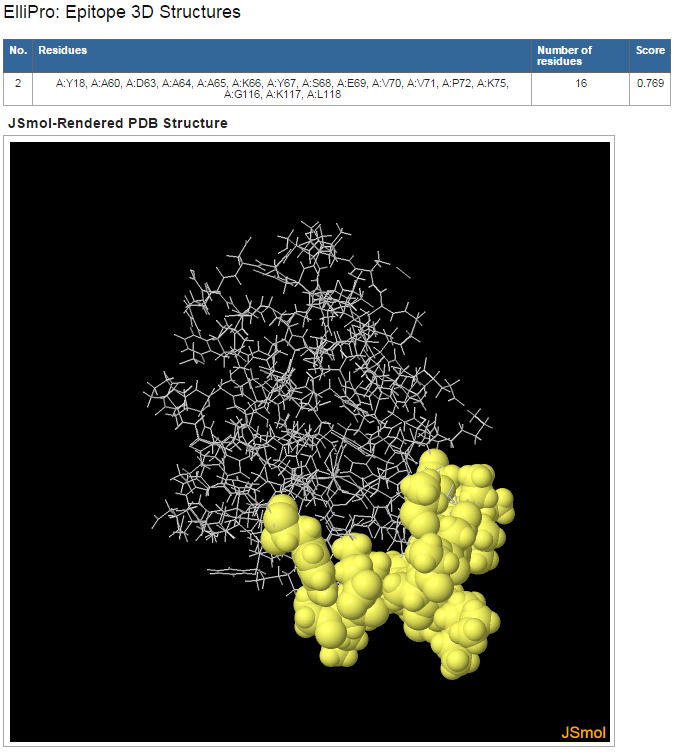
Figure 4. Example 3D structure mapping of a predicted discontinuous epitope
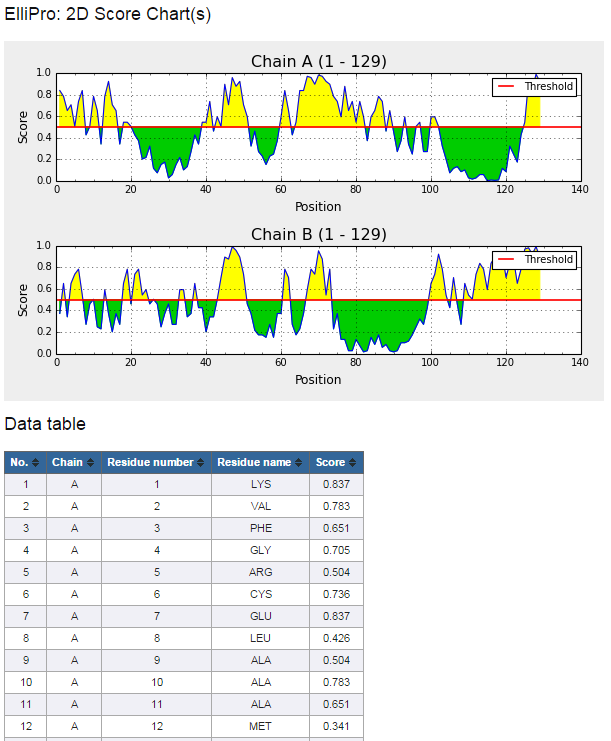
Figure 5. Example 2D score chart
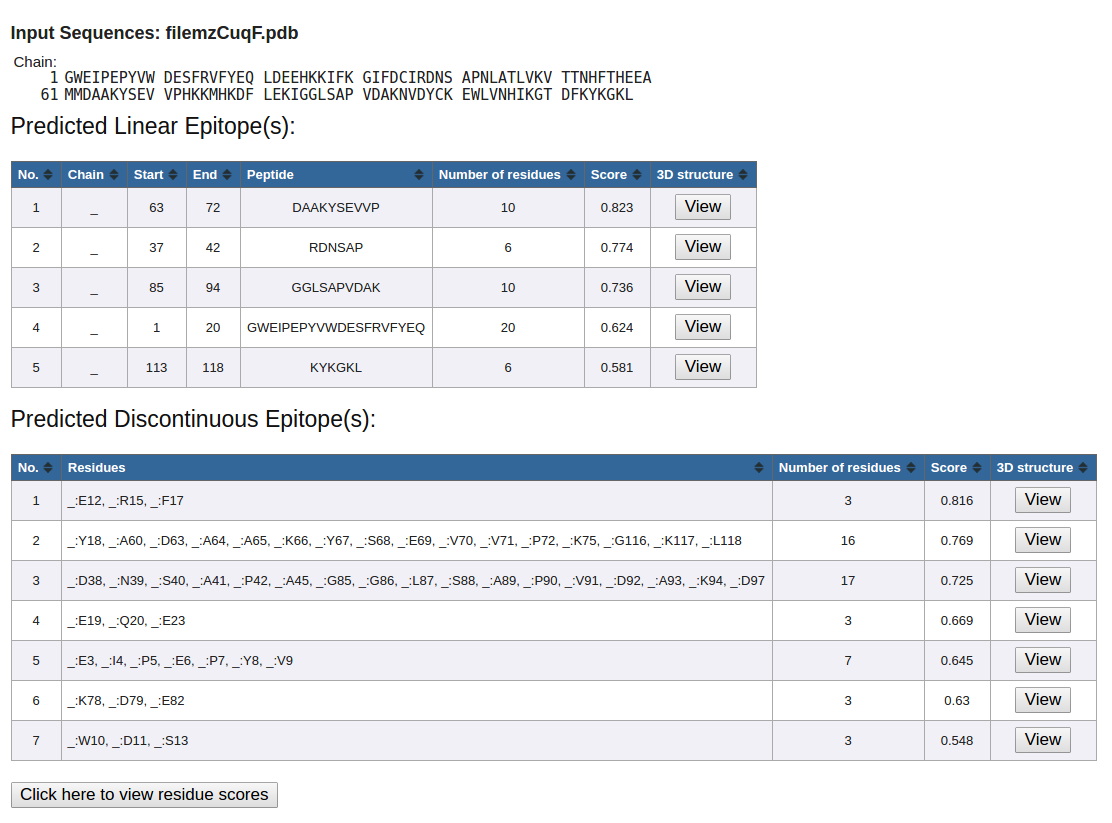
Figure 6. Example underscore as a placeholder
The results show:
- Protein Sequence(s)
- Predicted Linear Epitope(s)
- Predicted Discontinuous Epitope(s)
- Click on "View" in the "3D Structure" column following each predicted eptiope to view its 3D structure mapping (Figure 4)
- Click on "Click here to view residue scores" button at the bottom of the prediction result page (Figure 3) to view 2D score chart(s) of the protein sequence(s) (Figure 5)
- If there is no 'chain id' specified in the 'ATOM' records of the upload PDB file, underscore ('_') will be used as a placeholder. (Figure 6)
Figure 3. Example prediction results
Figure 4. Example 3D structure mapping of a predicted discontinuous epitope
Figure 5. Example 2D score chart
Figure 6. Example underscore as a placeholder