The Tepitool provides prediction of peptides binding to MHC class I and class II molecules. This page describes the tool in detail. For complete details please refer Paul, S., Sidney, J., Sette, A., and Peters, B. 2016. TepiTool: A pipeline for computational prediction of T cell epitope candidates. Curr. Protoc. Immunol. 114:18.19.1-18.19.24 (Journal link, PMID: 27479659 ).
The Tepitool is designed as a wizard with 6 steps as described below. Each field (except sequences and alleles) is filled with default recommended settings for prediction and selection of optimum peptides. The input parameters can be adjusted as per your specific needs. You can go back to previous steps to change your selection before submission of the job. Once you submit the job (at the end of step-6), you will not be able to make any more changes and will have to start the prediction all over again with updated input parameters.
Steps:
- Provide sequence data
- Select the host species and MHC allele class
- Select the alleles for prediction
- Select peptides to be included in prediction
- Select preferred methods for binding prediction and peptide selection strategy and cutoff values
- Review selections, enter job details and submit data
Step-1. Enter sequence data
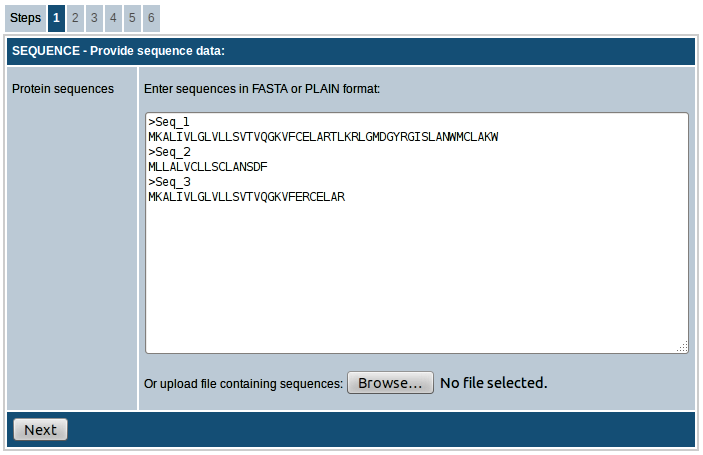
In the first step the sequences you want to scan for MHC binding peptides have to be entered. The sequences can either be entered directly into the text area provided or uploaded as a text file in FASTA or PLAIN format. In FASTA format a sequence begins with a single-line description, followed by lines of sequence data. The description line should start with a greater than symbol (">"). Please click here for more details on FASTA format. If ">" symbol is not found, the sequences will be considered to be in PLAIN format and each line will be considered a separate sequence. All sequences have to be amino acids specified in single letter code (ACDEFGHIKLMNPQRSTVWY). Example of FASTA formatted sequences:
>Seq_1
MKALIVLGLVLLSVTVQGKVFCELARTLKRLGMDGYRGISLANWMCLAKW
>Seq_2
MLLALVCLLSCLANSDF
>Seq_3
MKALIVLGLVLLSVTVQGKVFERCELAR
Step-2. Select the host species and MHC allele class
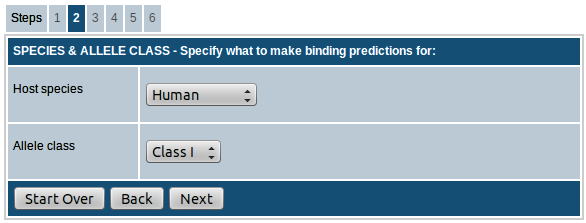
Here you can choose the species for which binding prediction has to be performed. You can choose from human, chimpanzee, cow, gorilla, macaque, mouse and pig.
For human and mouse, you have the option to choose between MHC class I and class II. For all other species, only class I is available for prediction.
Step-3. Select the alleles for prediction
In this step you can choose the alleles for prediction.
MHC class I
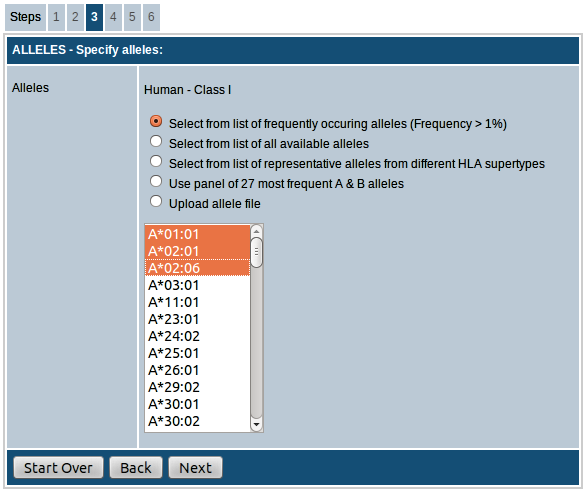
For all species you have the option of either choosing the alleles from the list of available
alleles provided or upload a list of alleles in the specified format as a text file.
For human, there are additional options to choose alleles. Descriptions of the options available for human are given below:
- Select from a list of frequently occurring alleles (frequency > 1%): This list provides the alleles with frequency > 1% in the global population. Follow this link for more information on
HLA allele frequencies.
- Select from list of all available alleles: This list provides all alleles for which we have prediction algorithm available.
- Select from list of representative alleles from different HLA supertypes: This list provides a set of representative alleles from MHC class I supertypes.
- Select panel of 27 most frequent alleles: This list contains the 27 most frequent alleles in the global population.
- Upload allele file: Upload text file with your choice of alleles in a plain text file. Alleles should be listed as one allele per row.
Sample format is given below. Follow this link for more information on nomenclature of HLA alleles.
Format:
A*01:01
A*01:02
A*01:03
MHC class II
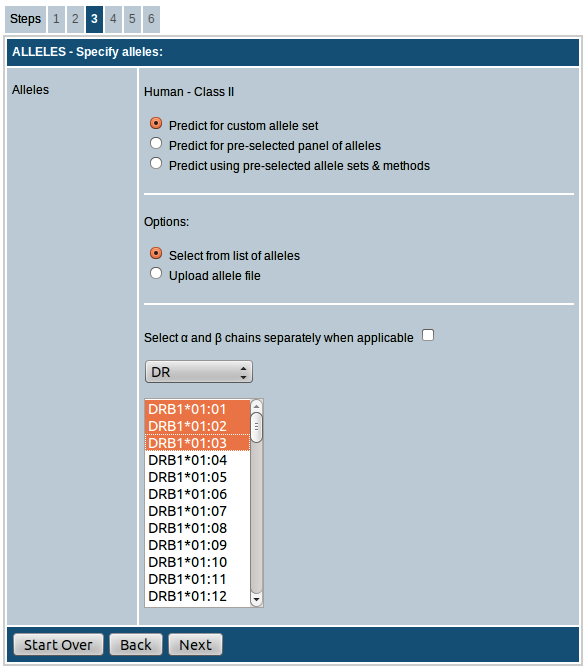
MHC class II is available for only human and mouse. For mouse, the options available are either choosing the alleles from the list of available
alleles provided or upload a list of alleles in the specified format as a text file.
Options for choosing alleles for human are described in detail below:
- Predict for custom allele set: This option allows you to choose your own alleles for prediction. Here you can select desired alleles from the list of available alleles provided separately for DP, DQ and DR loci. By clicking the checkbox provided you can choose α and β chains separately for DP and DQ alleles (this option is not available for DR alleles since only DR-β chain is effectively variable). You can also upload the list of alleles as a plain text file. Alleles should be listed as one allele per row.
Sample format is given below.
Format:
DRB1*01:01
DRB4*01:01
DQA1*01:01/DQB1*05:01
DQA1*05:01/DQB1*03:01
- Select for pre-selected panel of alleles: This option currently provides a set of 26 most frequent human class II alleles from DP, DQ and DR loci. You can choose alleles from each locus separately or any combination of the three loci.
- Predict using pre-selected allele sets & methods: This option is for prediction based on peptide's ability to bind to multiple alleles (called as "promiscuous binding"). This option uses pre-defined sets of alleles. There are two options for prediction based on promiscuous binding:
- Use the "7-allele method" for top immunodominant peptides: This option does prediction based on the median of the consensus percentile ranks for 7 DR alleles (DRB1*03:01, DRB1*07:01, DRB1*15:01, DRB3*01:01, DRB3*02:02, DRB4*01:01, DRB5*01:01). Peptides with median consensus percentile rank ≤ 20.0 are selected as predicted binders (this cutoff can be adjusted in step-6). More details are available in Paul et al., 2015
- Use panel of 26 most frequent alleles for promiscuous binding: In this option, the prediction is done based on the number of alleles binding to the peptide out of a set of 26 alleles that are most frequent in the global population. Consensus percentile rank of 20.0 is used as cutoff for binding to the allele and 50% alleles is chosen by default as the cutoff for number of alleles binding to determine binders.
Step-4. Choose peptides to be included in prediction
In this step you have the option to define parameters to provide an approximate number and nature of peptides to be included in the prediction.
MHC class I
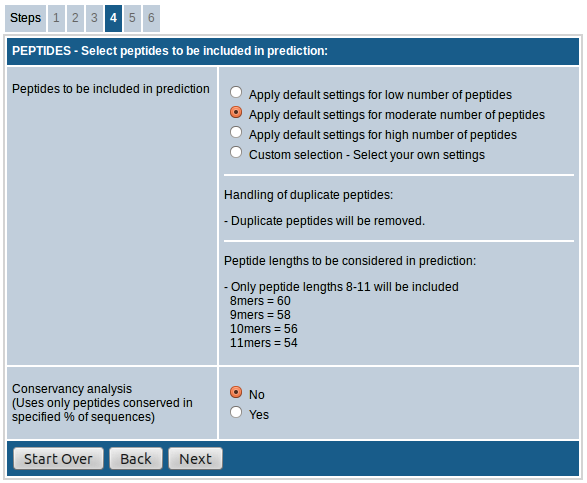
The first field, "Select peptides to be included in prediction”, provides the option to choose either default settings for low/moderate/high number of peptides or custom select the peptides to be included in the prediction which in turn determines the number of peptides in the prediction results.
- In case of option "low number of peptides”, only 9mer peptides are included and duplicate peptides are removed.
- When the option "moderate number of peptides” is chosen 8mers, 9mers, 10mers and 11mers are included and duplicate peptides are removed.
- The option "high number of peptides” includes all possible peptides from 8mers to 14mers and retains duplicate peptides.
- If "custom selection” is chosen you can adjust the handling of duplicate peptides and select peptide lengths as desired.
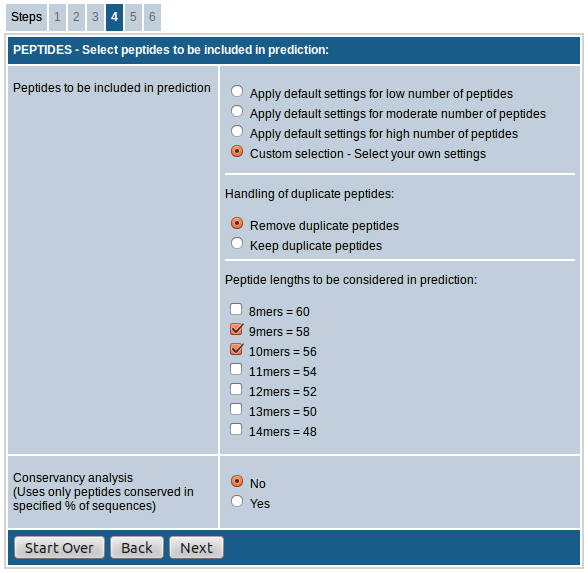
The option, "Handling of duplicate peptides", allows you to remove or retain identical peptides from different sequences in the result. That is, if a peptide sequence meeting the input parameters is present in more than one input sequence, the peptide will be included in the results only once if the "Remove duplicate peptides" option is chosen whereas it will be included as many times if "Keep duplicate peptides" option is chosen instead.
The option, "Peptide lengths to be considered in prediction", provides the option to choose the peptide lengths to be included in the prediction. For all species you have the option to choose from 8mers to 14mers. This field also shows the approximate number of peptides that will be included in the prediction process against each length.
The last field "Conservancy analysis" allows you to select only the peptides that are conserved in specified % of input sequences. This is useful especially in case of homologous sequences where you want to pick only the most conserved peptides. You also have the option to choose the desired % of sequences as cutoff if you opt to do the conservancy analysis.
MHC class II
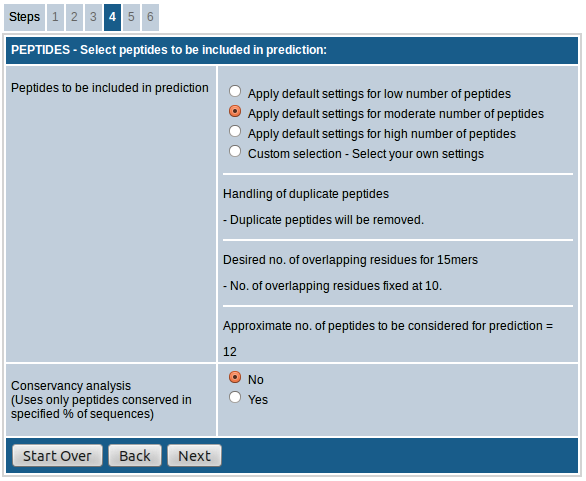
The options available for MHC class II are mostly the same as for MHC class I. One major difference is that in case of MHC class II the peptide length is fixed at 15. MHC class II molecules have an open binding groove (compared to the closed binding groove of MHC class I molecules) and can accommodate longer peptides compared to class I. However, the majority of the energy of the interaction between a peptide and a class II molecule is provided by a peptide core of about 9 amino acids in length. The presence of additional amino acids flanking the binding core also appears to be necessary for stable binding, although they do not specifically interact with the MHC peptide-binding groove. Accordingly, in common practice peptides of 15 residues are used for MHC class II binding predictions. Selecting 15mer peptides overlapping by 10 amino acid residues will provide the minimum number of peptides with all possible 9mer binding cores that has at least one flanking residue on both sides. In this way the possibility of selecting redundant peptides with the same binding core can be avoided.
The first field, "Select peptides to be included in prediction”, provides the option of choosing either default settings for low/moderate/high number of peptides or custom select the peptides to be included in prediction.
- In case of option "low number of predicted peptides”, the 15mers generated are overlapping by 8 AA residues and duplicate peptides are removed.
- The option "moderate number of peptides” increases the number of peptides included by increasing the number of overlapping residues to 10. The duplicate peptides are removed here as well.
- When the option "high number of peptides” is chosen, the number of overlapping residues remains same at 10, but duplicate peptides are retained.
- If "custom selection” is chosen you can adjust the handling of duplicate peptides and no. of overlapping residues as desired.
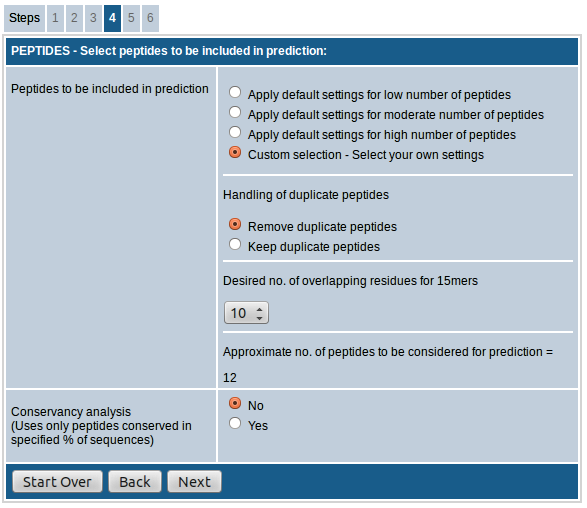
The option, "Handling of duplicate peptides", allows you to remove or retain identical peptides from different sequences in the result. That is, if a peptide sequence meeting the input parameters is present in more than one input sequence, the peptide will be included in the results only once if the "Remove duplicate peptides" option is chosen whereas it will be included as many times if "Keep duplicate peptides" option is chosen instead.
The option "Desired no. of overlapping residues for 15mers" lets you choose the number of overlapping residues for the 15mers to be generated. You can choose any number between 0-14. As the no. of overlapping residues increases, the number of peptides included in prediction increases proportionately.
The last section of this field shows the "approximate no. of peptides to be considered for prediction" based on the selections made above.
The last field "Conservancy analysis" allows you to select only the peptides that are conserved in specified % of input sequences. This is useful especially in case of homologous sequences. You also have the option to choose the desired % of sequences as cutoff if you opt to do the conservancy analysis.
The above options are available only if you select "predict for custom allele set" or "predict for pre-selected panel of alleles" option in step-3. If you select "Predict pre-selected allele sets and methods" in step-3, the duplicate peptides will always be removed and number of overlapping residues will be fixed at 10.
Step-5. Select prediction method, peptide selection strategy & cutoff values
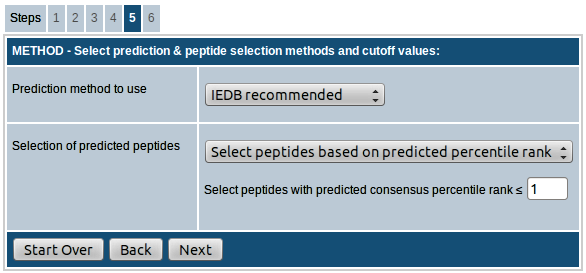
MHC class I
Here the first field provides you the option to choose the binding prediction method. The prediction method list box allows choosing from a number of MHC class I binding prediction methods: IEDB recommended, Consensus, NetMHCpan (version 2.8), ANN (Artificial neural network, also called as NetMHC, version 3.4), SMM (Stabilized matrix method), SMMPMBEC (SMM with a Peptide:MHC Binding Energy Covariance matrix) and Comblib_Sidney2008 (Scoring Matrices derived from Combinatorial Peptide Libraries).
IEDB recommended is the default prediction method selection. Based on availability of predictors and previously observed predictive performance, this selection tries to use the best possible method for a given MHC molecule. Currently for peptide:MHC-I binding prediction, for a given MHC molecule, IEDB Recommended uses the Consensus method consisting of ANN, SMM, and CombLib if any corresponding predictor is available for the molecule. Otherwise, NetMHCpan is used. This choice was motivated by the expected predictive performance of the methods in decreasing order: Consensus > ANN > SMM > NetMHCpan > CombLib.
Of note, we fully expect the IEDB recommendation to change as we perform larger benchmarks of newly developed methods on blind datasets to determine an accurate assessment of prediction quality. Towards that end, automated benchmarks have been established to continuously evaluate the performance of the existing MHC class I binding methods. These benchmarks are updated weekly as new datasets are deposited into the IEDB and the latest results can be found here.
The second field provides options of different approaches for selection of predicted peptides. There are five different ways you can choose the predicted peptides:
- Select peptides based on predicted percentile rank: In this approach, the peptides will be selected based on the predicted percentile rank. You have the option to provide the cutoff percentile rank for determining binders. The default value provided is 1.0, i.e. all peptides with percentile rank ≤ 1.0 will be selected as predicted peptides.
- Select peptides based on predicted IC50: Here the peptide selection is based on the predicted IC50 nM. You have the option to provide the cutoff IC50 value for determining binders. The default value provided is 500 nM, i.e. all peptides with predicted IC50 ≤ 500 nM will be selected as predicted peptides. Please note that if this approach is chosen, the prediction method will always be NetMHCpan irrespective of the prediction method chosen in the first filed.
- Select peptides based on MHC specific predicted binding threshold (Applicable to human only): In this option the peptides are selected based on allele specific binding thresholds as described in Paul et al., 2013. The threshold for each allele has been defined in a way as the selected peptides will account for 75% of the total immune response. Please note that this option is available for only a certain number of human alleles as listed on the option field in step-5.
- Select top x% of predicted peptides: This option allows you to simply select the specified % of peptides out of all peptides included in prediction. For example, if you want to select top 5% 9mers and there are approximately 100 9mers in all, this approach will select the top 5 9mers as predicted peptides. The peptide selection criterion in this approach will always be predicted percentile rank.
- Select top x number of predicted peptides: This option is same as above, the difference being that here you can input the exact number of peptides you want instead of the %. Here also the peptide selection criterion is always predicted percentile rank.
MHC class II
Similar to MHC class I, the first field provides you the option to choose the binding prediction method. The prediction method list box allows choosing from a number of MHC class II binding prediction methods: IEDB recommended, Consensus, NetMHCIIpan (version 3.0), NN_align (Artificial neural network, also called as NetMHCII-2.2, version 2.2), SMM_align (also called as NetMHCII-1.1, version 1.1), Sturniolo and Combinatorial library (Scoring Matrices derived from Combinatorial Peptide Libraries).
IEDB recommended is the default prediction method selection. Based on availability of predictors and previously observed predictive performance, this selection tries to use the best possible method for a given MHC molecule. Currently for peptide:MHC-II binding prediction, for a given MHC molecule, IEDB Recommended uses the Consensus method consisting of NN_align, SMM_align and Sturniolo/Combinatorial Library if any corresponding predictor is available for the molecule. Otherwise, NetMHCIIpan is used. The Consensus approach considers a combination of any three of the four methods, if available, where Sturniolo is a final choice. The expected predictive performances are based on large scale evaluations of the performance of the MHC class II binding predictions: a 2008 study based on over 10,000 binding affinities, a 2010 study based on over 40,000 binding affinities and a 2012 study comparing pan-specific methods. Supplementary information for evaluation of predictive tools are available for 2008 and 2010 studies.
Of note, we fully expect the IEDB recommendation to change as we perform larger benchmarks of newly developed methods on blind datasets to determine an accurate assessment of prediction quality.
The second field provides options of different approaches for selection of predicted peptides. There are five different ways you can choose the predicted peptides:
- Select peptides based on predicted percentile rank: In this approach, the peptides will be selected based on the predicted percentile rank. You have the option to provide the cutoff percentile rank for determining binders. The default value provided is 10.0, i.e. all peptides with percentile rank ≤ 10.0 will be selected as predicted peptides.
- Select peptides based on predicted IC50: Here the peptide selection is based on the predicted IC50 nM. You have the option to provide the cutoff percentile rank for determining binders. The default value provided is 500 nM, i.e. all peptides with predicted IC50 ≤ 1000 nM will be selected as predicted peptides. Please note that if this approach is chosen, the prediction method will always be NetMHCIIpan irrespective of what you choose in the first filed.
- Select peptides based on the number of alleles binding: This approach is based on peptide's ability to bind to multiple alleles (called as "promiscuous binding") and is same as the second option provided in step-3 except that here you can select your own set of alleles. Percentile rank of 20.0 is used as cutoff for binding to the allele and 50% alleles is chosen by default as the cutoff for number of alleles binding to determine binders. You have the option to adjust the latter cutoff.
- Select top x% of predicted peptides: This option allows you to simply select the specified % of peptides out of all the peptides generated. For example, if you want to select top 5% 9mers and there are approximately 100 9mers in all, this approach will select the top 5 9mers as predicted peptides. The peptide selection criterion in this approach will always be predicted percentile rank.
- Select top x number of predicted peptides: This option is same as above, the difference being that here you can input the exact number of peptides you want instead of the %. Here also the peptide selection criterion in this approach will always be predicted percentile rank.
The above options are available only if you select "Predict for specific allele(s)" option in step-3. If you select "Predict promiscuous binding" in step-3, the prediction method will always be "IEDB recommended". If the "7-allele method" is chosen, the selection of predicted peptides is based on the median consensus percentile rank of the 7 alleles. All peptides with median consensus percentile rank ≤ 20.0 will be selected. You have the option to adjust this cutoff value. If the promiscuous binding based on the 26 most frequent alleles is chosen, the peptide selection is based on the number of alleles binding, i.e. peptides that bind to at least 50% of alleles (13 alleles) will be selected.
Step-6. Review summary, enter job details & submit data
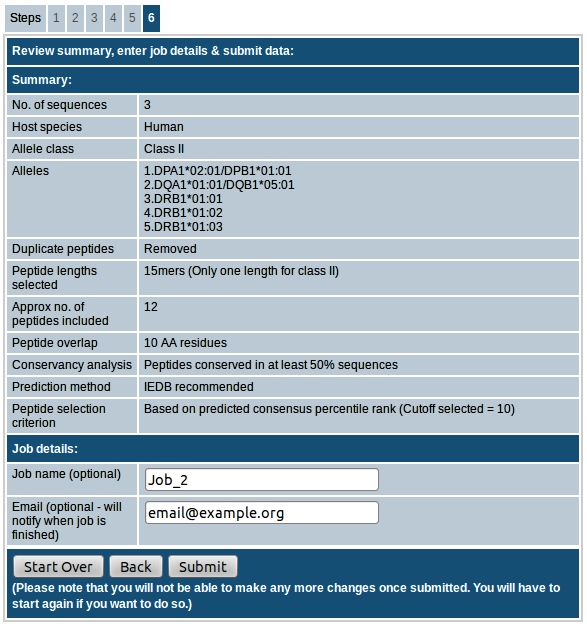
In the last step you can review the summary of input parameters you provided. You also have the option to provide a job name and email id. Providing job name will make it easy for us to troubleshoot in case anything goes wrong. If email id is provided, an email will be sent with the concise results once the job is finished. If you want to make any changes, you can go back to previous steps and edit as needed. Please note that once you submit the job, you will not be able to make any more changes and you will have to start the prediction all over again if you want to do so. Please do not close or refresh the browser window until the results are obtained.
Results
The prediction results page has the following sections:
- Concise results
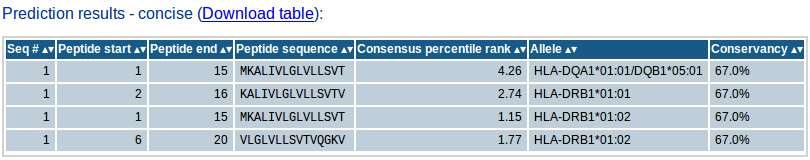
The concise results table shows the final list of predicted peptides selected based on the input parameters provided. The table will contain the sequence # of the peptide's source protein in the input sequence set, start and end positions of the peptide within the source protein sequence, the peptide sequence, the selection criterion parameter value (percentile rank/IC50/median consensus percentile rank/number of binding alleles), the allele (where applicable) and conservancy % (if conservancy analysis is done). If conservancy analysis is done, the concise result will also show the conservancy of each peptide within the input sequence set. The concise results table can also be downloaded as csv file which can be opened using any spreadsheet such as MS Excel for further analysis. This section will be included in the email.
- Download results details

This section provides links for downloading the results details as csv files. It can include the following based on the input parameters:
- Non-redundant results (applicable only to Class II): This file will contain prediction results with redundant peptides within each sequence removed. Redundant peptides means peptides that overlap with more number of residues than desired. This result set includes both positive and negative peptides based on the input parameters.
- Complete results: This file will contain binding predictions of all peptides. This will include the predicted IC50 and percentile rank or other scores depending on the prediction method chosen. In case of IEDB recommended or consensus method, the results will include details from each of the prediction methods employed.
- Conservancy of peptides (applicable only if conservancy analysis is done): This file will contain conservancy of each peptide in the input sequence set.
- Citation information

This section contains details of references based on the prediction methods used that should be cited if you use the prediction results in your study. This section will also be included in the email.
- Input sequences

This section contains the input sequence set along with the sequence titles that you provided for binding prediction. If no titles were included for sequences (e.g. in case of plain sequence format), automatic sequence title will be provided in the format of ">Seq_1" etc.
- Other input parameters
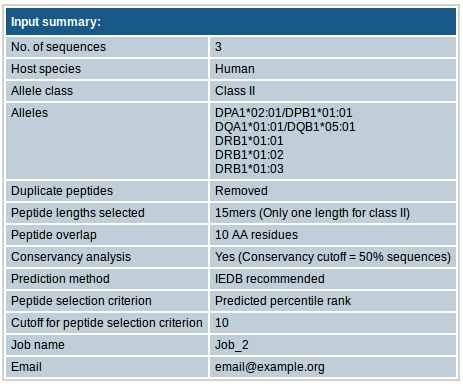
This section contains all other input parameters that you selected.
Interpretation of results
The concise results show the selected peptides that came top based on the binding predictions as per the input parameters you provided. In cases of peptide selection based on percentile rank, median consensus percentile rank (7-allele method) and IC50, lower value indicates better predicted binding affinity. In case of promiscuous binding based on number of alleles, predicted binding affinity is better when the number of binding alleles is higher. The recommended input parameters for prediction and selection of optimum peptides are provided as default input parameters.
Each of the binding prediction algorithms predicts the binding affinity in terms of IC50 nM. A percentile rank is generated by comparing the peptide’s predicted binding affinity against that of a large set of similarly sized peptides that are randomly selected from the SWISS-PROT database. Percentile scores provide a uniform scale allowing comparisons across different predictors. A lower percentile rank indicates higher affinity. In the case of the consensus method, the median percentile rank of the three methods involved is used to generate the consensus percentile rank. While the output of the predictions is quantitative, there are systematic deviations from experimental IC50 values. For example, the makeup of the training data and the prediction methods used have a non-trivial impact on the range of predicted IC50 values.