Lymphocyte Receptor Automated Modelling (LYRA) - Tutorial
- One continuous sequence
- FASTA format
All sequences have to be amino acids specified in single letter code
(
ACDEFGHIKLMNPQRSTVWY)
Example:
sequence for light chain here...
>heavy chain header
sequence for heavy chain here...

HMMER:
HMMER is used for searching sequence databases for sequence homologs, and for making sequence alignments.
HMMER + SCWRL 4.0:
HMMER is used for searching sequence databases for sequence homologs, and for making sequence alignments. Non conserved residues side chains are modeled using SCWRL 4.0. SCWRL uses conserved residues side chains as constraints when modeling remaining side chains. This is the default choice.
1DEE,8FAB,1fve) to explicitly
prevent lyra to use them as framework or loops template for both chains.
The Summary table shows template structures and calculated canonical structures for both chains.
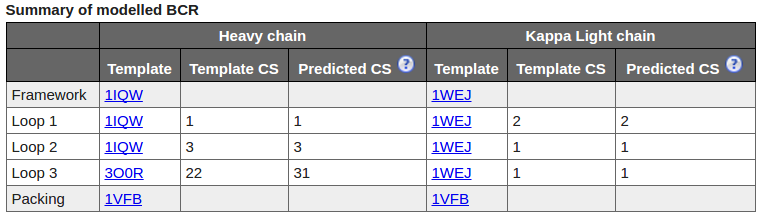
Canonical Structures(CS)
Shown below is the definitions of the canonical structures for TCR chains. Structures are predicted using the
affinity propagation algoritm, and the exemplars are shown as being the representative PDB structure.
|
|
BCR Canonical structures are predicted using sequence-based rules. The rules are described in the following table:
|
|
|
Alignment: The sequences are aligned with the input sequence, the template sequence and the variable loops.

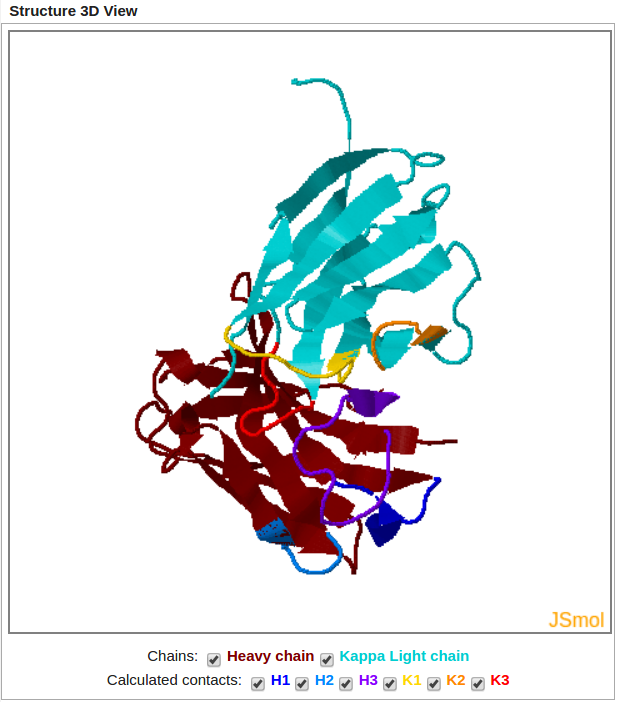
Modeled Structure is displayed using JSmol Viewer (JavaScript-Based Molecular Viewer From Jmol).
It allows the user to display or hide both chains and to highlight each loop.
Click on Download button to download the model generated by LYRA.
The downloaded model file can be visualized locally using Jmol viewer. Jmol is a free, open source molecule viewer that is cross-platform.
Instructions on how to download, install and run the viewer can be found here.