MHC-NP - Tutorial
How to obtain predictions
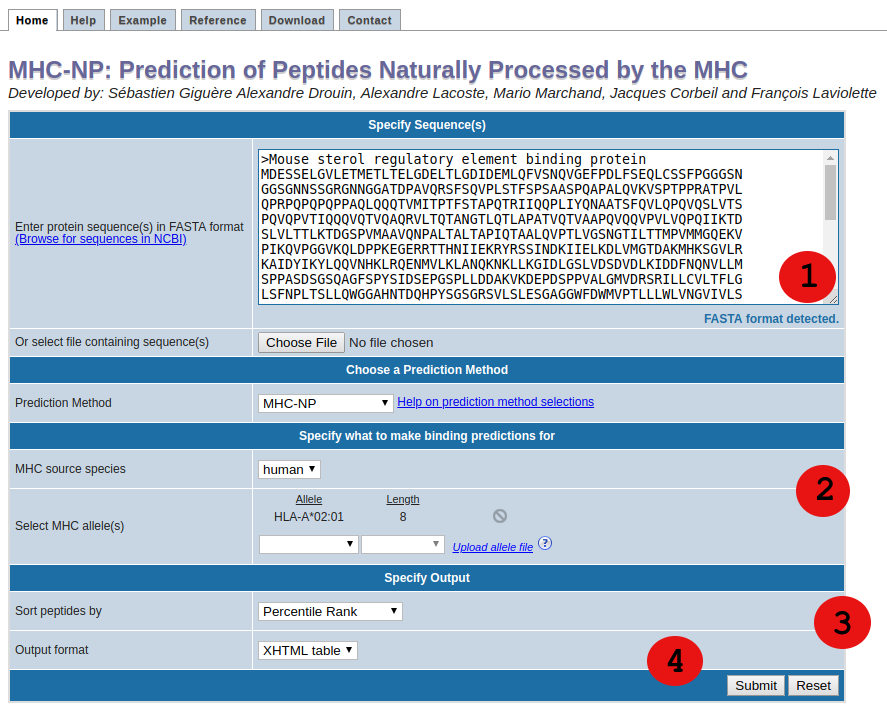
1. Specify sequences
First, specify the sequences you want to scan for naturally processed peptides. The
sequences should either be entered directly into the text area field labeled "Enter protein
sequence(s)”, or can be taken from a file that has to be uploaded using the "Browse"
button. Please enter no more than 200 FASTA sequences or upload file size less than or
equal to 10 MB per query.
The sequences can be supplied in three different formats:
All sequences have to be amino acids specified in single letter code
(
- Space separated sequences
- One continuous sequence
- FASTA format
All sequences have to be amino acids specified in single letter code
(
ACDEFGHIKLMNPQRSTVWY)
2. Specify method and what to make predictions for
MHC-NP tool provides two different methods to make predictions.
- MHC-NP
- NetMHCpan EL
Predictions are not limited to peptides of one specific length binding to one specific allele,
but multiple allele/length pairs can be submitted at a time. The allele / peptide length
combination can be selected using the list boxes in this section.
• Format for the upload allele file:
File should be in simple text format containing comma separated values, where each allele is separated from it's length by a comma followed by a new line.
Therefore, you can upload a file with each line containing only one such allele-length pair(example given below). However, you may also
choose allele(s) and their length(s) from the drop-down selection in together with your uploaded file.
Example:
Example:
H-2-Db,9
HLA-B*07:02,9
HLA-A*02:01,10
...
Additional information regarding HLA allele frequencies
and nomenclature are also provided.
HLA-B*07:02,9
HLA-A*02:01,10
...
3. Specify the output
The menus in this section change how the prediction output is displayed.
By default, the predictions are displayed in an XHTML table. From the results page, this table can be downloaded as a commaseparatedvalue (CSV) file. In addition, to reuse the prediction results in an external program, it is possible to retrieve the predictions in a plain text format. To do this, choose "Text file" in the output format listbox.
By default, the predictions are displayed in an XHTML table. From the results page, this table can be downloaded as a commaseparatedvalue (CSV) file. In addition, to reuse the prediction results in an external program, it is possible to retrieve the predictions in a plain text format. To do this, choose "Text file" in the output format listbox.
4. Submit the prediction
This one is easy. Click the submit button, and a result screen similar to the one below should appear.
Interpreting prediction output
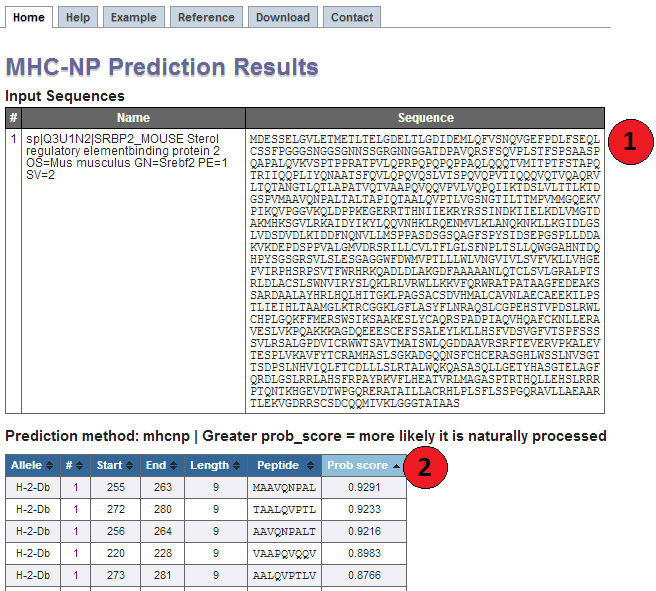
1. Input Sequences
This table displays the sequences and their names extracted from the user input. If no
names were assigned by the user (which is only possible in FASTA format), the
sequences are numbered in their input order (sequence 1, sequence 2, ...).
2. Prediction output table
Each row in this table corresponds to one prediction. The columns contain the allele the
prediction was made for, the input sequence number (#), start position and end position of the peptide,
its length, the peptide and the predicted probability score. The table can be sorted by clicking on the table
column headers.
For each peptide, a score is predicted. This score is included between 0 and 1 and the greater the value, the more likely a peptide is naturally processed by a specific allele. When looking at the results, you should consider the ranking of a peptide compared with other peptides for which predictions were made.
Knowing how MHCNP was trained can help with the interpretation of the predictions. The data that were used to train the tool were composed of 3 groups of peptides : Binders, Nonbinders and Eluted peptides (see http://bio.dfci.harvard.edu/DFRMLI/HTML/natural.php#training).

Eluted peptides were eluted from a MHC/peptide complex and are thus considered naturally processed peptides. To make predictions, MHCNP relies on a combination of two predictors. The first predictor was trained to distinguish binding peptides from nonbinding peptides for a given allele. The second predictor was trained to distinguish eluted peptides from other binding peptides.
Thus, a given peptide can fall in 4 distinct groups. MHCNP aims to predict naturally processed peptides, therefore we focus on predicting peptides that fall in the upperleft cell of the table below. The score that is outputted is an estimation of the probability that the peptide falls in this cell of the table. The greater the score, the more likely the peptide is naturally processed by the MHC allele.
For more information see the paper about MHCNP in the reference section.
For each peptide, a score is predicted. This score is included between 0 and 1 and the greater the value, the more likely a peptide is naturally processed by a specific allele. When looking at the results, you should consider the ranking of a peptide compared with other peptides for which predictions were made.
Knowing how MHCNP was trained can help with the interpretation of the predictions. The data that were used to train the tool were composed of 3 groups of peptides : Binders, Nonbinders and Eluted peptides (see http://bio.dfci.harvard.edu/DFRMLI/HTML/natural.php#training).
Eluted peptides were eluted from a MHC/peptide complex and are thus considered naturally processed peptides. To make predictions, MHCNP relies on a combination of two predictors. The first predictor was trained to distinguish binding peptides from nonbinding peptides for a given allele. The second predictor was trained to distinguish eluted peptides from other binding peptides.
Thus, a given peptide can fall in 4 distinct groups. MHCNP aims to predict naturally processed peptides, therefore we focus on predicting peptides that fall in the upperleft cell of the table below. The score that is outputted is an estimation of the probability that the peptide falls in this cell of the table. The greater the score, the more likely the peptide is naturally processed by the MHC allele.
For more information see the paper about MHCNP in the reference section.
The original developer of MHC-NP tool are
Sébastien Giguère and
Alexandre Drouin.