Deimmunization predictions - Tutorial
The immunogenic regions are predicted using broad spectrum alleles (26 reference alleles) for MHC class II in human population. The server will make the 15mer which overlaps in 10 residues. Each generated 15mer will be predicted for 26 alleles using IEDB recommended method. The IEDB recommended is a consensus method from three best methods available for an allele. You can find the details of MHC class II reference alleles and recommended method in Class II prediction tool help page.
1. The input sequence can be pasted in the text area of the form.
2. The second input is to choose a threshold.
The threshold correspond to median percentile rank for each peptide (15mer window with an overlap of 10mer) from IEDB recommended method in 26 reference alleles in MHC class II.
Lower the percentile rank, higher the binding affinity of a peptide to a wide range of alleles.
Therefore, seleting the higher threshols for percentile rank, will turn the more number of peptides as immunogenic in next step. The default is set to 20, which is our recommendations.
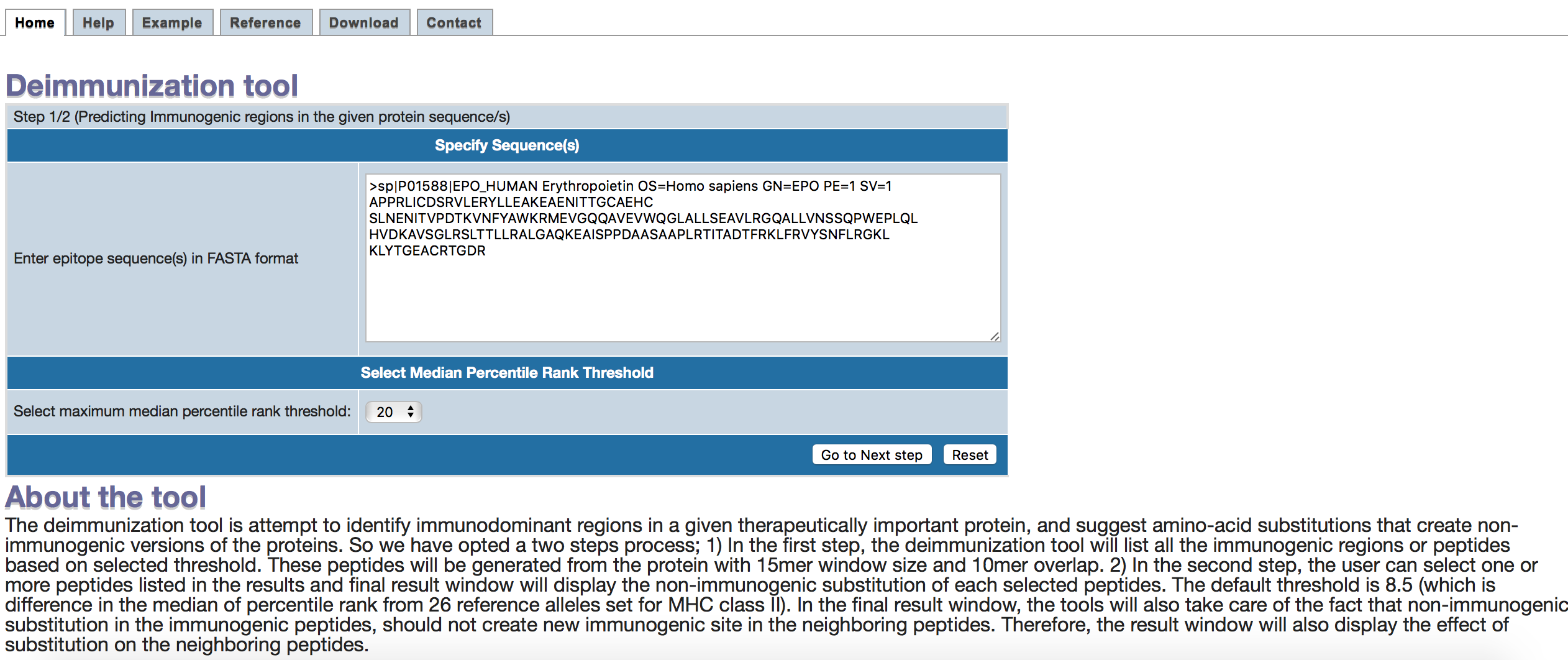
The step 2 (intermediate) page will display all the immunogenic peptides predicted in the user provided sequence. These peptides will then subjected to all (285) the possible mutants for a 15mer peptides. The non-immunogenic peptides should have higher median percentile rank for immunogenicity. In our study, we found that 8.5 higer is optimum for deimmunization and we set it as default. User can change the threshold, if he/she wish to do so.
1. User has an option to select multiple peptides. Check the boxes for each peptide selection. One peptide has to be selected for running the server.
2. The threshold for the difference in median percentile rank can be changed. Defult is 8.5.
3. The job name is optional.
4. We recommend to provide email address,so that you can receive the results. As the server may take some time to make predition for 285 variants of each selected peptides, predicted for 26 alleles and 3 methods in IEDB recommended method.
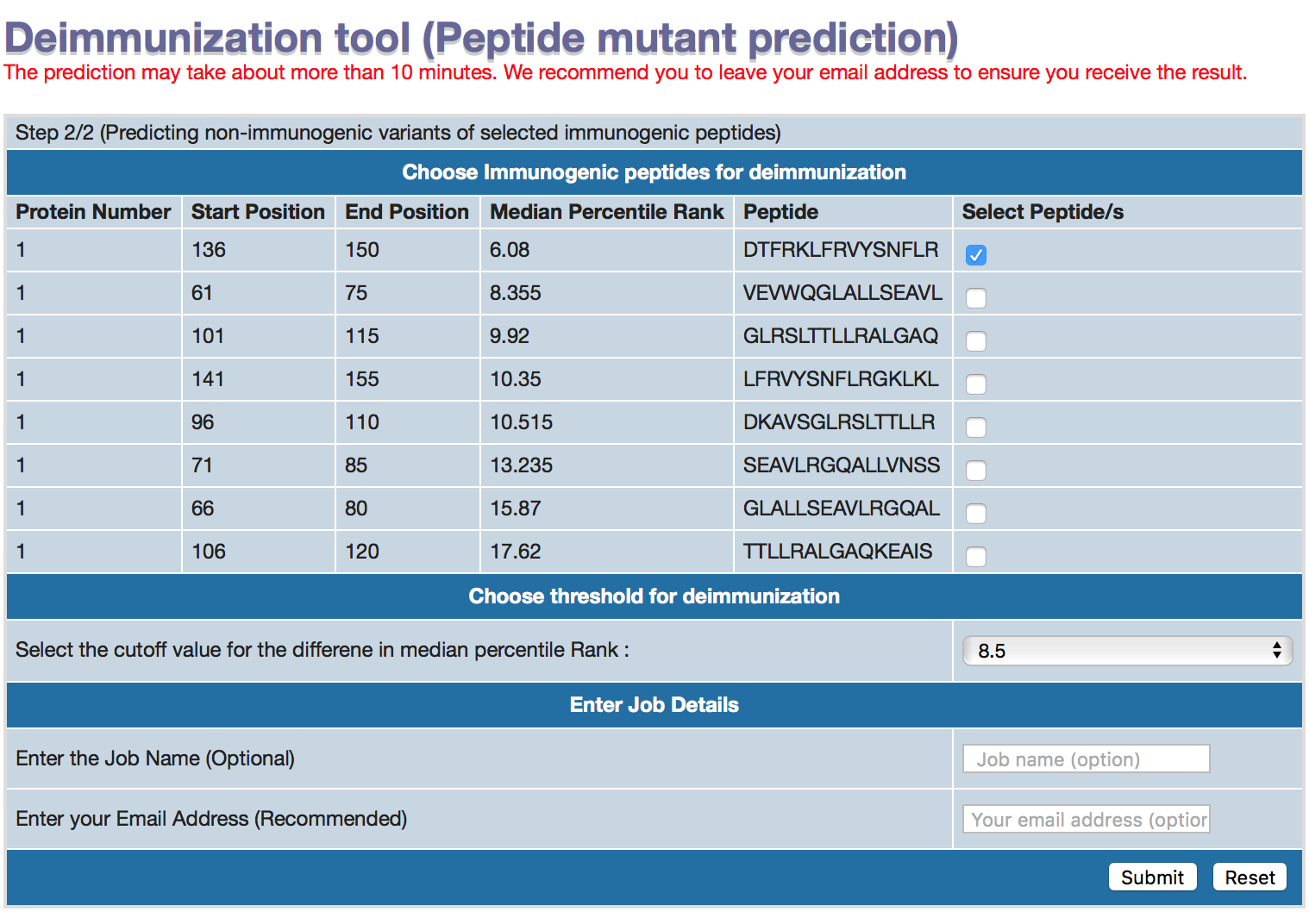
The table header are names in such way to be interpreted easily. The protein number is the serial number of protein in the given sequences at first step.
The median percentile rank is the median for percentile rank in "IEDB reommended method" for 26 alleles with broad coverage of human population.
Higher the difference in Median percentile rank denotes the low binding affinities.
The default is set 8.5 according to our recommendation.
This means, all the mutants of a selected peptide with percentile rank = 'wild type median percentile rank + selected median percentile threshod' will be displayed in the result page.
User can also selecte to show all the mutants or all the less immunogenic mutants.
The neigbhoring effect is summarized in Deimmunization score
Deimmunization score has a range of 1-10.
The score is 1, when an user submit a peptide of 15 residue. In this case there would be no neighbor to evaluate.
The score will be 10, when both the neighbors has become immunogenic with that particular mutation.
Here is the table for score details.
| Immunogenicity of Neighboring peptide (1) | Immunogenicity of Neighboring peptide (2) | Score |
| Absent | Absent | 1 |
| Reduced | Absent | 2 |
| Reduced | Reduced | 3 |
| Neutral | Absent | 4 |
| Reduced | Neutral | 5 |
| Neutral | Neutral | 6 |
| Increased | Absent | 7 |
| Reduced | Increased | 8 |
| Increased | Neutral | 9 |
| Increased | Increased | 10 |
The short interpretation has been given on the result page. It clearly says that Higher the median differnce will make the peptide less immunogenic and Lower Immunogenic score is preferred for minimum effect on neighboring peptides.